ISO 13485 2016 Package
2025-03-27 11:30ISO 13485 2016 Package
Get your accreditation at the lowest possible cost


ISO/IEC 13845:2016 complete package
2016 version

Price : 489 $
Certification Made Simple and Accessible for Your Business
The complete ISO/IEC 13485 2016 package is a comprehensive document package that contains everything from all the templates of procedures, processes, forms, checklists, tools, detailed guides, and instructions needed to:
- Start your ISO/IEC 13485 process.
- Create your ISO/IEC 13485 documentation.
- Quickly access ISO/IEC 13485 accreditation.
- Benefit from an ISO/IEC 13485 management system that is simple and adapted to the needs of your organization.

Why start with a blank page. Start your Project TODAY, and save up to 80% on your time and money.

This package comes with 1 hour Live 1-to-1 Online Session with ISO consultant, document reviews, continual email support for 12 months and regular update service.

Cost-Effective Implementation: Much cheaper than an on-site consultant, and requires much less time than doing it from scratch
ISO/IEC 13845 2016 Version Complete Package
• Added Value: All ISO/IEC 13485 2016 requirements have been developed into an efficient process that adds operational value to your organization and consequently increases productivity.
• Effective: Minimal effort is required to follow procedures necessary to meet all requirements of ISO/IEC 13485 2016.
• Simplified: Bureaucracy and excessive paperwork have been eliminated from each process to make it easy—while remaining fully compliant with ISO/IEC 13485 2016.
Start your Project TODAY, and save up to 80% on your time and money.
The all-in-one document package for ISO/IEC 13845 2016 version
Save time, save money and simplify the accreditation process.
Documents included:

Forms
This package provides you with the following features:
- Full lifetime access
- Access on a laptop, desktop, and mobile
Certificate of completion
This Package Includes
Procedures:
- Quality Manual
- Document Control Procedure
- Record Control Procedure
- Management Review Procedure
- Internal Audit Procedure
- Corrective Action Procedure
- Preventive Action Procedure
- Risk Management Procedure
- Design and Development Procedure
- Purchasing Procedure
- Supplier Evaluation Procedure
- Receiving Inspection Procedure
- Production Control Procedure
- Calibration Procedure
- Maintenance Procedure
- Training Procedure
- Nonconforming Product Procedure
- Complaint Handling Procedure
- Advisory Notice Procedure
- Regulatory Reporting Procedure
- CAPA Procedure
Manual and quality policy
• Quality manual
SOPs
- SOP for Quality System Maintenance
- SOP for Change Management
- SOP for Labeling and Packaging
- SOP for Product Storage and Distribution
- SOP for Traceability
- SOP for Validation and Verification Activities
- SOP for Installation and Servicing
- SOP for Sterilization Process Control (if applicable)
- SOP for Cleanroom Procedures (if applicable)
- SOP for Software Validation (if applicable)
- SOP for Post-Market Surveillance
- SOP for Medical Device Reporting
- SOP for Cybersecurity Management (if applicable)
- SOP for Environmental Monitoring (if applicable)
- SOP for Product Return and Recall

ISO 13485 Documentation Requirements Explained
The ISO 13485 Documentation Package is a vital resource for medical device manufacturers and related organizations that need to establish a robust Quality Management System (QMS). This comprehensive package includes all the essential documentation required to comply with ISO 13485:2016—the international standard for quality management systems in the medical device industry.
Comprehensive and well-structured documentation is crucial for ensuring product safety, regulatory compliance, and consistent manufacturing processes. From design control to post-market surveillance, this package helps streamline your path to certification and maintain trust with regulators and customers.
Why ISO 13485:2016 Documentation Matters
ISO 13485:2016 is specifically designed for organizations involved in the lifecycle of medical devices. Proper documentation ensures product quality, risk management, traceability, and regulatory compliance across all stages—from development to distribution. It also serves as a foundation for inspections, audits, and global market access.
This package includes:
Key ISO 13485:2016 Documentation Categories
| Document | Purpose in Quality Management System |
|---|---|
| Quality Manual | Describes the scope, structure, and processes of the QMS |
| Medical Device File | Contains detailed information about each medical device |
| Design and Development Documentation | Controls and records product design activities and changes |
| Risk Management File | Identifies, evaluates, and controls risks throughout the product lifecycle |
| Validation and Verification Records | Confirms that processes and products meet specified requirements |
| Supplier Evaluation and Monitoring | Ensures the quality and reliability of purchased products and services |
| Complaint Handling and CAPA Procedures | Manages customer feedback and drives corrective and preventive actions |
| Post-Market Surveillance Records | Tracks product performance and safety after release |
Core ISO 13485:2016 Documentation Requirements
Management System Documentation
To comply with ISO 13485:2016, organizations must implement a documented QMS that ensures medical device quality and regulatory alignment. This package includes:
Quality Policy and Objectives – Defines the organization’s commitment to product quality and regulatory compliance.
Document Control and Record Retention Procedures – Ensures documentation accuracy, accessibility, and security.
Internal Audit Plans and Reports – Evaluates QMS effectiveness and identifies areas for improvement.
Management Review Meeting Minutes – Documents leadership’s review and strategic decisions for the QMS.
Training and Competence Records – Tracks employee qualifications and ongoing training activities.
Product Realization and Operational Documentation
These documents help manage the complete lifecycle of medical devices, ensuring safe and compliant product delivery. This package includes:
Design and Development Plans – Outlines project timelines, responsibilities, and design inputs/outputs.
Production and Process Control Procedures – Ensures consistency and control in manufacturing operations.
Installation and Servicing Instructions – Provides standardized guidance for equipment setup and maintenance.
Process Validation Protocols – Demonstrates that critical processes consistently produce expected results.
Identification and Traceability Records – Tracks components and final products for recall and compliance purposes.
Monitoring, Feedback, and Improvement Documentation
Continuous improvement is essential in a regulated environment. This package includes:
Nonconformity and Corrective Action Reports – Documents deviations and the actions taken to resolve them.
Customer Feedback and Complaint Records – Captures field data and supports responsiveness to quality issues.
Risk Analysis and Updates – Ensures risk evaluations stay current and integrated into product decisions.
CAPA (Corrective and Preventive Action) Logs – Provides evidence of systemic improvement efforts.
Product Recall Procedures – Ensures swift and effective actions in the event of product withdrawal.
Legal and Regulatory Documentation
Regulatory Compliance Matrix – Maps applicable regulations and how they are addressed within the QMS.
Technical File and Device Master Records – Maintains complete documentation required for product approval.
Supplier Quality Agreements – Defines roles, responsibilities, and quality expectations with suppliers.
Confidentiality and Data Protection Policies – Ensures compliance with privacy laws and data handling standards.
Ensure Compliance with ISO 13485:2016 Today!
Achieving ISO 13485:2016 certification can be demanding, but the ISO 13485 Documentation Package provides everything your organization needs to build and maintain a compliant, effective, and audit-ready Quality Management System. With ready-to-use templates and detailed records, you’ll be prepared to meet both customer expectations and regulatory requirements.
💡 Get started today and take your medical device QMS to the next level with a structured, compliant, and ISO 13485:2016-ready documentation system
90 Days Money Back Guarantee

If for whatever reason during the FIRST 90 days of your purchase, you are not satisfied for any reason, simply contact support@qse-academy.com and our support team will issue you an immediate and full refund.
The package includes all the documents you need to comply with ISO/IEC 13845 2016 – these documents are fully acceptable by the accreditation audit.
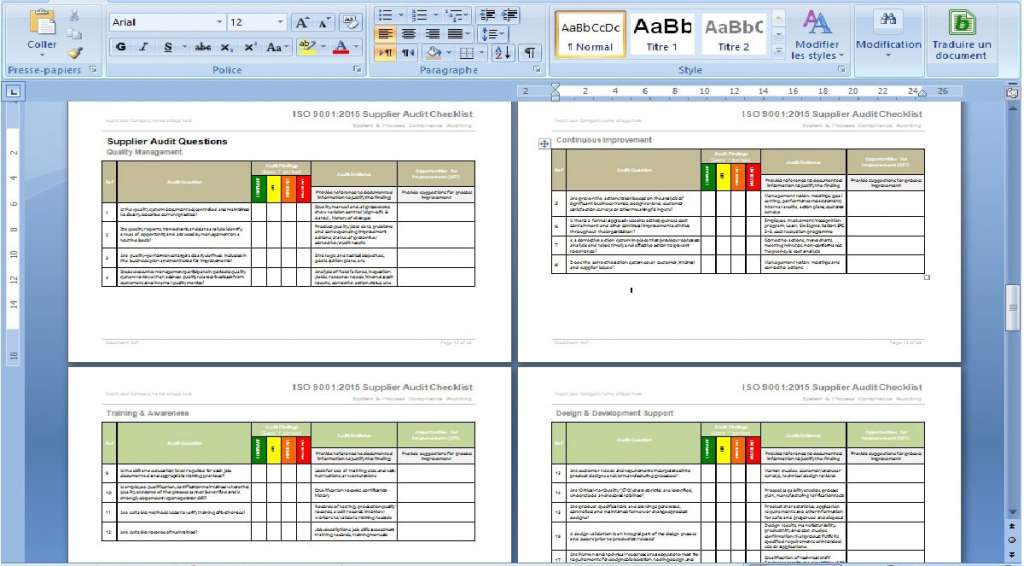
All documents are in MS Word or MS Excel, to make them very easy to customize for your business. You can customize them by adding company logos and colors, and edit headers and footers to match your favorite style.
We have already completed about 90% of the information requested on the documents. To complete them you must fill in only the name of the company, the responsible parties, and any other information unique to your company. you will be guided through the process, commenting on the elements that are needed and those that are optional.
We presented the ISO 13845 documentation, so as to assure all its users that they have completed everything accurately and with the utmost efficiency.

All the documents are made so that you can follow the proposed order perfectly, which allows you to make sure that nothing is missing, and that no one gets lost in the process.
The included comments and flowcharts help your staff understand each document and its usefulness, which helps you to make quality management more fluid, and processes easier to follow.
Features of the complete ISO/IEC 13845 2016 Kit
Price: 489 $
– Documentation included: 58 documents for the implementation of ISO 13845
– MS Office 2007 format, MS Office 2010, MS Office 2013
– Language: English
– Documents are fully editable – just enter the information specific to your business.
– Acceptable for the ISO 13845 2016 accreditation audit? Yes, all the documents required by ISO 13845 2016 are included, as well as the quality policy and the current but optional procedures.
Instant Delivery – The package is downloadable immediately after purchase
Free Consultation – In addition, you can submit two complete documents for review by professionals.
Created for your business – The models are optimized for small and medium businesses.

Complete ISO/IEC 13845 2016 Package
The complete kit to implement ISO/IEC 13845
Price : 489 $
Total Implementation Duration: 8 Months
ISO/IEC 13845 Implementation Project Plan
Achieving ISO 13845 is a significant milestone for any organization, signifying a commitment to data protection and privacy. Our expert consultants are here to guide you through every step of the implementation process, from initial consultation and gap analysis to final assessment and compliance certification. With our comprehensive project plan, tailored training programs, and dedicated support, we ensure your organization meets all ISO 13845 requirements efficiently and effectively. Partner with us to enhance your organization’s credibility, improve data handling processes, and gain trust on an international scale. Let us help you achieve excellence in data privacy management.
Introduction: Project Kick-off and Gap Analysis (Duration: 1 Month)
Introductory Tasks
1.1 ISO 13485 Kick-off and Awareness
Task: Organize Kick-off Meeting
- Description: Conduct a kick-off meeting to introduce the ISO 13485:2016 project to key stakeholders, discussing objectives, scope, timelines, and responsibilities.
- Deliverables: Project plan, meeting agenda, and minutes.
- Meeting: Initial consultation with senior management and implementation team.
1.2 Perform Gap Analysis
Task: Conduct Gap Analysis Against ISO 13485:2016 Requirements
- Description: Assess the organization’s current QMS and processes against ISO 13485:2016 requirements to identify gaps.
- Deliverables: Gap analysis report with identified non-conformities.
- Meeting: Review findings with management and the quality team.
Section 1: Quality Management System (QMS) Planning (Duration: 2 Months)
2.1 Develop and Document Quality Policy and Objectives
Task: Define Quality Policy and Objectives
- Description: Develop a quality policy and measurable quality objectives that align with ISO 13485:2016 and the organization’s strategic direction.
- Deliverables: Documented quality policy and objectives.
- Meeting: Review with senior management for approval.
2.2 Develop QMS Documentation
Task: Create QMS Documentation (Procedures, Work Instructions)
- Description: Document all key processes required by ISO 13485, including production, design, risk management, document control, and training.
- Deliverables: QMS manual, procedures, work instructions.
- Meeting: Review documented procedures with the QMS team.
Section 2: Risk Management and Regulatory Compliance (Duration: 2 Months)
3.1 Implement Risk Management Processes (ISO 14971 Integration)
Task: Identify and Document Risk Management Procedures
- Description: Develop and implement risk management procedures based on ISO 14971 (Application of Risk Management to Medical Devices) to ensure the identification, analysis, and control of risks associated with medical devices.
- Deliverables: Risk management plan and procedure.
- Meeting: Review risk management plan with the design and production teams.
3.2 Ensure Regulatory Compliance (ISO 13485 Clause 7.2)
Task: Define Regulatory Requirements for Medical Devices
- Description: Identify all applicable regulatory requirements related to medical devices in the relevant markets (e.g., FDA, CE marking) and ensure they are integrated into the QMS.
- Deliverables: Regulatory compliance matrix.
- Meeting: Review regulatory requirements with the regulatory affairs and quality assurance teams.
Section 3: Product Realization and Design Controls (Duration: 1 Month)
4.1 Establish Product Design and Development Processes (ISO 13485 Clause 7.3)
Task: Develop Design and Development Procedures
- Description: Establish procedures for the design and development of medical devices, including design planning, inputs, outputs, verification, validation, and design reviews.
- Deliverables: Design and development procedure, design review templates.
- Meeting: Review with engineering and design teams.
4.2 Implement Design Change Controls
Task: Establish a Design Change Management Process
- Description: Create a design change control process to ensure that all changes are documented, validated, and reviewed for their impact on product safety, performance, and regulatory compliance.
- Deliverables: Design change control procedure.
- Meeting: Review with design, production, and quality teams.
Section 4: Supplier Management and Traceability (Duration: 1 Month)
5.1 Supplier Evaluation and Approval (ISO 13485 Clause 7.4)
Task: Develop Supplier Evaluation Procedures
- Description: Implement a supplier evaluation and approval process to ensure that suppliers meet the required quality standards and regulatory requirements.
- Deliverables: Supplier evaluation criteria, approved supplier list.
- Meeting: Review supplier performance and approval criteria with procurement.
5.2 Implement Traceability and Control of Medical Devices
Task: Develop Traceability Procedures (ISO 13485 Clause 7.5)
- Description: Establish a traceability system to ensure that the history, application, and location of medical devices can be traced throughout production, delivery, and installation.
- Deliverables: Traceability procedure, traceability records.
- Meeting: Train staff on traceability procedures and conduct system testing.
Section 5: Control of Non-Conforming Product and Corrective Actions (Duration: 1 Month)
6.1 Control of Non-Conforming Product (ISO 13485 Clause 8.3)
Task: Establish Procedures for Handling Non-Conforming Products
- Description: Develop procedures to identify, document, segregate, and dispose of non-conforming products, ensuring they do not reach customers or patients.
- Deliverables: Non-conformance reports, corrective action logs.
- Meeting: Review non-conformance procedures with production and quality teams.
6.2 Implement Corrective and Preventive Actions (ISO 13485 Clause 8.5)
Task: Develop Corrective Action Procedures
- Description: Implement a corrective and preventive action (CAPA) process to investigate the root cause of non-conformities and take preventive actions to avoid recurrence.
- Deliverables: CAPA reports, root cause analysis documentation.
- Meeting: CAPA review meeting with senior management and department heads.
Section 6: Internal Audits and Management Review (Duration: 2 Months)
7.1 Internal Audit Program (ISO 13485 Clause 8.2.4)
Task: Develop an Internal Audit Plan
- Description: Create an internal audit program to evaluate the effectiveness of the QMS, covering all areas required by ISO 13485:2016, including design, production, supplier management, and regulatory compliance.
- Deliverables: Internal audit plan, audit checklist.
- Meeting: Audit planning meeting with the audit team and quality manager.
7.2 Conduct Internal Audits
Task: Perform Internal Audits
- Description: Conduct internal audits to verify compliance with the QMS and ISO 13485 requirements. Identify any non-conformities and areas for improvement.
- Deliverables: Internal audit reports, non-conformance reports.
- Meeting: Post-audit review meeting with the management team to discuss findings and corrective actions.
7.3 Conduct Management Review (ISO 13485 Clause 5.6)
Task: Schedule and Conduct a Management Review
- Description: Conduct a management review to assess the effectiveness of the QMS and its alignment with business objectives. Ensure top management evaluates QMS performance and opportunities for improvement.
- Deliverables: Management review meeting minutes, action plans.
- Meeting: Present results and discuss strategic improvement actions with senior management.
Final Assessment: Certification Audit Preparation and External Audit (Duration: 1 Month)
8.1 Pre-Certification Audit
Task: Perform a Pre-Certification Internal Audit
- Description: Conduct a pre-certification audit to verify that the QMS meets ISO 13485:2016 requirements and is ready for the certification audit. Address any remaining non-conformities.
- Deliverables: Pre-certification audit report, corrective action plan.
- Meeting: Final review with senior management to confirm readiness for the external audit.
8.2 Certification Body Selection and External Audit
Task: Select Certification Body and Schedule Certification Audit
- Description: Select an accredited certification body for the ISO 13485 audit. Coordinate the external audit schedule and ensure the organization is fully prepared.
- Deliverables: Certification body selection report, external audit schedule.
- Meeting: Final meeting with management and quality team to confirm readiness for the certification audit.
This 8-month project plan for ISO 13485:2016 implementation provides a structured approach to achieving certification. The plan covers the key areas of quality management, risk management, design and development controls, supplier management, non-conformance handling, and internal audits, culminating in the final certification audit.
What our customers think:

“We were initially overwhelmed by the thought of getting ISO 13485 accreditation, but the QSE Academy package made everything so much easier. The documents were well-organized, and about 90% of the work was already done for us! We only had to customize minor details, which saved our team a lot of time and effort. We particularly appreciated the 1-on-1 online session with the ISO expert, which helped us navigate tricky parts of the accreditation. This package saved us both time and money compared to hiring external consultants.”
Sarah M.
Compliance Manager

“Implementing ISO 13485 seemed like a daunting task at first, but the package from QSE Academy was a game-changer for us. Not only did it streamline the entire process with pre-built templates and guidelines, but it also saved us considerable expenses by eliminating the need for full-time consultants. The support was fantastic, and we were able to complete the project in a fraction of the time we initially estimated. Highly recommend this for anyone looking for a cost-effective way to get accredited!”
James K.
Operation Director
Frequently Asked Questions
How long will it take to receive the complete package of documents after I place my order?
Upon completing your purchase, you will be redirected to the download page immediately. Additionally, a link to access your file will be sent to your email. The files are provided in a .zip format, which you will need to extract. If you encounter any issues with the download, please do not hesitate to contact us at support@qse-academy.com. Our support team is always ready to assist you.
What payment methods can I use?
We offer several payment options for your convenience. You can choose to pay using a credit card, debit card, or PayPal. Additionally, we provide a flexible layaway plan for those who prefer to pay for their purchase over time. If you have any questions about our payment options, please don’t hesitate to contact us.
Do you offer a money-back guarantee if I'm not satisfied with the service?
We offer a 30-day money-back guarantee. If you are not satisfied with our service for any reason, you can cancel within the first 30 days and receive a full refund, no questions asked.
Is there ongoing support or assistance available after my purchase?
Yes! At QSE Academy, our ISO experts provide continued support by answering your queries via email. You can expect a detailed response within 24 to 48 hours to help you move forward confidently.
Are updates to the documentation package included after purchase?
Absolutely. To ensure your documentation remains reliable and compliant, we update our packages every 6 months. Existing customers receive these minor updates at no extra charge. However, when there’s a major revision of the ISO standard itself, you’ll need to purchase an updated kit to align with the new standard.
Will I receive a valid invoice for my business expenses after completing the purchase?
Yes. After completing your purchase, you’ll immediately receive a valid invoice suitable for business and tax purposes. If you require any specific adjustments or details added to your invoice, please reach out to our support team.
Can I customize these documents for my company's specific needs?
Yes, the documents are fully customizable! You can easily edit, modify, and add your company’s logo to tailor them specifically for your organization. Additionally, if you’d prefer assistance, we offer a personalized “Done-For-You” customization service to deliver audit-ready documents tailored exactly to your organization’s requirements.
How quickly can I implement this ISO standard using your documentation?
Implementation time varies depending on your company’s engagement, resources, and experience. Typically, we’ve observed businesses successfully achieve compliance and certification within 3 to 6 months using our clear, structured documentation packages.
Do these documents guarantee successful certification?
While our documentation packages significantly simplify the certification process, the ultimate success of ISO certification depends on effective implementation. For organizations seeking further assurance, we also provide comprehensive support services, including guided implementation and internal audits, to help you confidently pass your certification audit.
Do you offer hands-on assistance if I need extra help during implementation?
Definitely! If you prefer a complete, hands-off solution, we offer a premium “Done-For-You” implementation service. Our ISO experts handle the full preparation, providing you with audit-ready documentation and detailed implementation support. You simply adopt the customized materials, follow the tailored guidelines, and confidently pass your audit.



